
Cells are the building blocks of life and they perform a wide variety of functions for most living organisms. The innerworkings of a cell are driven in large part by cell organelles that each have specific functions and purposes. Organelles work together like organs in the human body to ensure the health and proper functioning of the cell. Lysosomes in particular garner less recognition and seem to be less understood by most microbiological enthusiasts.
Lysosomes are organelles formed in the golgi apparatus that are responsible for degrading foreign elements and internal molecules by employing acid hydrolases that break down materials that the cell no longer uses. Lysosomes are commonly referred to as the digestive system of the cell.
However, there is so much more to this organelle. In this post we will go through the structure, function, history, and so much more!
Lysosome Structure and Function
Lysosomes are membrane-enclosed organelles that contain acid hydrolases (hydrolytic enzymes that work on acidic conditions) capable of breaking down proteins, nucleic acids, carbohydrates and lipids. Lysosomes are therefore known as the digestive system of the cell and serve to degrade substances taken up from outside the cell and to digest elements the cell no longer uses.
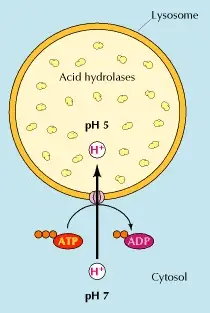
Lysosomes are described as cytoplasmic dense spherical bodies surrounded by a limiting membrane that can have a high variation in size and shape. Lysosomes contain around 50 degradative enzymes that can break down proteins, DNA, RNA, polysaccharides and lipids.
All these enzymes are acid hydrolases that function in an acidic pH of about 5. Lysosomes need to constantly concentrate protons to maintain their acidic pH. This is achieved thanks to a proton pump that uses energy in the form of ATP (Adenosine triphosphate) to transport protons from the cytosol into the lysosome (figure 1) (Cooper 2000, Saftig 2005).
Lysosome Formation
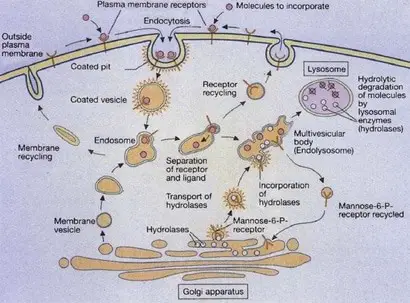
Lysosomes are formed by the fusion of clathrin-coated vesicles from the trans-Golgi network with late endosomes. Endosomes are intermediate stages in the formation of lysosomes. Early endosomes are located at the cell periphery, and contain molecules that are taken up by endocytosis (the process of incorporate external substances to the cell through vesicles), specifically ligand-receptors complexes (a ligand could be defined as a signaling molecule and a receptor as a receiving molecule).
Then the receptors are carried to the recycling endosomes (a system of tubular vesicles) and the ligands are translocated to late endosomes with a more acidic pH of 5.5. Lysosomal membranes and enzymes are packaged in the trans-Golgi network, enclosed in clathrin-coated vesicles and transported to late endosomes, forming endolysosomes, which then mature to lysosomes (figure 2)(Cooper 2000, Gartner & Hiatt 2012).
Intracellular Digestion
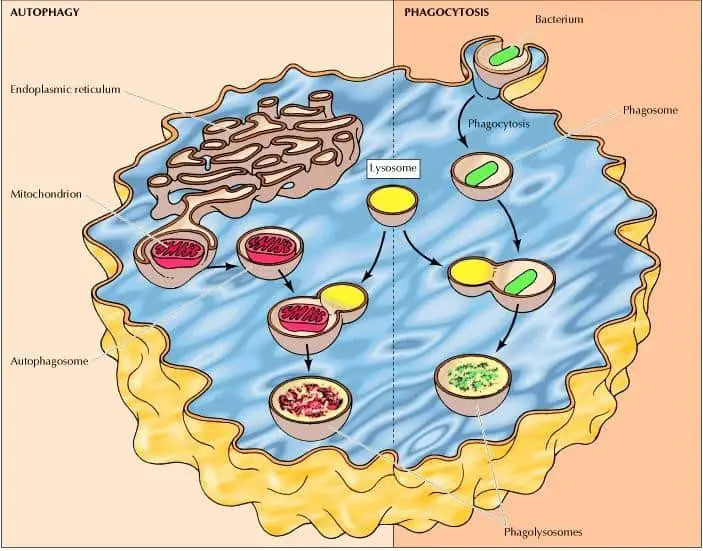
The intracellular degradation process led by lysosomes has three routes, the degradation of molecules taken up by receptor-mediated endocytosis, by phagocytosis and autophagy.
Phagocytosis is a process characterized by the scooping up and degradation of large particles (this includes bacteria), cell debris, and aged cells that need to be removed. These large particles are taken by phagosomes (phagocytic vacuoles), which then fuse with lysosomes forming phagolysosomes that are usually large and heterogeneous due to their size and shape is determined by the content that is being digested (figure 3) (Cooper 2000).
On the other hand, autophagy is a process characterized by the take-up and degradation of the cell’s own components. This begins with the enclosure of an organelle (e. g. mitochondria) by a membrane derived from the endoplasmic reticulum forming an autophagosome. These autophagosomes then fuse with lysosomes forming phagolysosomes where their contents are digested (figure 3) (Cooper 2000).
Lysosome History
In the late 19th century the first reports related to lysosomes surfaced. Élie Metchnikoff and other scientists were able to identify certain external molecules at the point they entered the cell and when they were later “digested” by the cell.
In the 1950s, Christian Duve and his colleagues standardized a protocol for the purification of lysosomes in their research on the mechanisms of insulin action involving glucose-6-phosphatase enzymes. They noticed that acid phosphatase activity could only be observed when membranes were disrupted.
After this discovery, Duve and coworkers proposed the name lysosomes as they contained several hydrolases. After lysosomes were named and more clearly understood, lysosomes began to be studied in relationship to cell storage diseases, characterized by cellular accumulations of molecules.
The first research that linked lysosomes with these diseases was led by Henri Hers in 1963 who found that a deficiency in the lysosome enzyme acid maltase caused a glycogen storage disease (Novikoff et al. 1965, Maxfield et al. 2016).
Lysosome Related Diseases
Lysosome storage diseases involve a hereditary deficiency that prevents the degradation of its contents. Mutations on the genes that decode these enzymes cause the accumulation of molecules within the lysosomes of affected individuals.
Currently more than 30 genetic diseases have been reported. Among the mutations related to lysosomes malfunction is the incapability of targeting lysosomal enzymes to the lysosomes. The deficiencies include the activator proteins (4 small nonenzymatic glycoproteins known as Sphingolipid Activator Proteins or SAPs and the GM2 Activator protein) (Table 1) that activate lysosomal hydrolases and the inability of transporting small molecules, such as amino acids, monosaccharides, cofactors, and cations, from the lysosome to the cytoplasm (Cooper 2000, Gartner & Hiatt 2012, Ferreira & Gahl 2017).
| Activator | Activated Enzyme | Disease |
|---|---|---|
| SAP A | β-Galactosylceramidase | Krabbe |
| SAP B | Arylsulfatase A α-Galactosidase | Metachromatic Leukodystrophy |
| SAP C | β-Glucosylceramidas | Gaucher |
| SAP D | Sphingomyelinase | Niemann-Pick |
| GM2 Activator Protein | β-Hexosaminidase A | GM2 AB variant |
Lysosomal storage disorders have been reported to affect 1 in every 5,000 births, although this number might be higher due to the lack of recognition that pathology is caused by deficiencies in lysosomes. These patients are usually normal at birth and develop the disease in the first year of life and their pathophysiological consequences include neurodegeneration, metabolic imbalance and growth severe retardation (Cooper 2000, Pryor 2012, Ferreira & Gahl 2017, Lawrence & Zoncu 2019).
Pompe Disease
Lysosomal storage diseases due to enzyme defects can be categorized based on the macromolecule that cannot be degraded and is consequently stored. Glycogen storage disease or Pompe disease is caused by the accumulation of carbohydrates due to a deficiency of the enzyme acid alpha-glucosidase that hydrolyzes glycogen.
This disease has a broad age spectrum, rate of progression, and extent of organ involvement, especially in tissues where lysosomal glycogen accumulates, such as skeletal, cardiac and smooth muscles.
Wolman Disease
Wolman disease and cholesteryl ester storage disease are caused by the accumulation of neutral lipids (triglycerides and cholesterol esters), these diseases are characterized by a deficiency of lysosomal acid lipases that hydrolyze triglycerides and cholesteryl esters.
Wolman disease is an infantile disorder where affected infants suffer from liver cirrhosis, pulmonary fibrosis and adrenal calcification and insufficiency consequence of the accumulation of the aforementioned lipids (Kishnani et al. 2006, Du et al. 2008, Ferreira & Gahl 2017).
Fucosidosis
Fucosidosis, α- and β-mannosidosis, sialidosis, and aspartylglucosaminuria (named by the sugar that is not hydrolyzed) are disorders caused by the accumulation of glycoproteins. Fucosidosis disorder causes severe neurological symptoms, mental retardation, recurrent infections, growth retardation and dysostosis multiplex (severe abnormalities in the development of skeletal cartilage and bone and mental retardation) due to the absence of the α-L-fucosidase enzyme activity.
Mannosidosis Disorder
α-mannosidosis disorder causes slowed growth, mental and motor deterioration and severe infections due to the deficiency of the α-mannosidase enzyme.
β-mannosidosis disorder causes mental retardation, low muscle tone, cutaneous lesions and hearing loss due to the deficiency of the β-mannosidase enzyme.
Sialidosis Disorder
Sialidosis disorder causes early infant forms of mental retardation, dysostosis multiplex, hepatosplenomegaly (swelling and enlargement of liver and spleen) and early death due to the decreased activity of glycoprotein sialidase enzyme.
Aspartylglucosaminuria disorder causes mental retardation, slow development, diarrhea, frequent respiratory infections and cutaneous lesions due to the deficiency of the aspartylglucosaminidase enzyme. Most of these diseases affect children and cause early death in severely affected patients (Greiner-Tollersrud & Berg 2013, Johnson 2015, Ferreira & Gahl 2017).
Niemann-Pick Disease
Among metabolic disorders caused by sphingolipid storage deficiencies are several that deserved to be mentioned. Niemann-Pick disease has different clinical severities, type A is characterized by neuron degeneration, enlargement of lymph nodes and ocular manifestations, few patients survive beyond the 4 years of age and often affects Eastern European Jewish, and type B is similar to type A but it doesn’t affect the central nervous system and can cause spleen enlargement (Ferreira & Gahl 2017).
Gaucher’s Disease
Gaucher’s disease is another sphingolipid disorder and the most common lysosomal storage disease characterized by a failure of lysosome normal functioning which results in the accumulation of non-degraded elements and the following increase in the size and number of lysosomes within the cell.
This disorder results in cellular malfunction and pathological consequences to affected organs, for instance, neurological symptoms and the liver and spleen enlargement. This disease is specifically caused by the deficiency of glucocerebrosidase, a lysosomal enzyme responsible for catalyzing the hydrolysis of glucocerebroside to glucose and ceramide (Cooper 2000).
Other Lysosome Related Diseases
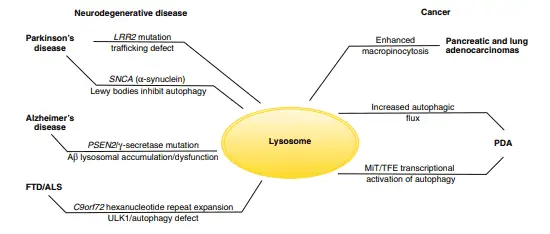
Lysosomes malfunction is also involved in the development of neurodegenerative diseases, such as Parkinson’s disease, Alzheimer’s disease and Huntington’s disease. The reason why these diseases are related to lysosomes is that there are some post-mitotic cells (mature cells that can’t undergo mitosis), such as neurons and cardiac myocytes, that depend on lysosomes to remove waste materials. Studies made on mice found that genetic inactivation of autophagy in the central nervous system results in spontaneous neurodegeneration in the absence of other genetic alterations that also cause these diseases.
Parkinson’s disease is caused both by a mutation that leads to the formation of insoluble aggregates in the α-synuclein protein (known as Lewy bodies) that inhibit autophagy, and a mutation that regulates the endomembrane trafficking, causing a general disruption of trafficking pathways and inhibition of autophagy.
On the other hand, Alzheimer’s disease is caused by a mutation that promotes the accumulation of cleaved β-amyloid peptides resulting in the inability to perform autophagy. Also, hereditary frontotemporal dementia and amyotrophic lateral sclerosis are caused by the formation of ubiquitin-positive intracellular inclusions that generates defects in the autophagy process.
Diverse mutations that prevent the correct function of autophagy are also related to several other neurodegenerative diseases, such as Huntington’s disease, spinocerebellar ataxia type 3, and variants of frontotemporal dementia and amyotrophic lateral sclerosis (figure 4) (Maxfield et al. 2016, Lawrence & Zoncu 2019).
Lysosomes also play an important role in human cancers. Autophagy is essential in the growth and survival of pancreatic and lung adenocarcinomas, which are cancers that have the ability to grow in microenvironments with poor oxygen and nutrients and satisfy their demand for metabolites by recycling intracellular and extracellular components.
This results in micropinocytosis, which involves the massive absorption of extracellular components degraded in the lysosome and allowing, in consequence, the growth of pancreatic ductal adenocarcinoma by breaking down macromolecules into amino acids.
The opposite process can also promote the growth of pancreatic ductal adenocarcinoma as the autophagic breakdown of intracellular proteins and ribosomes produces amino acids and nucleotides, this enhanced activity is led by the MiT/TFE factors that act at the transcriptional level (figure 4) (Lawrence & Zoncu 2019).
The treatment of lysosomal storage diseases is only safe and efficacious for very few cases. Even when the therapy is available early detection is vital before irreversible changes can occur, this applies to the bone marrow transplantation used to treat some enzyme deficiencies and cystinosis (another storage disease caused by the accumulation of the amino acid cystine) (Ferreira & Gahl 2017).
Lysosomes and Cell Death
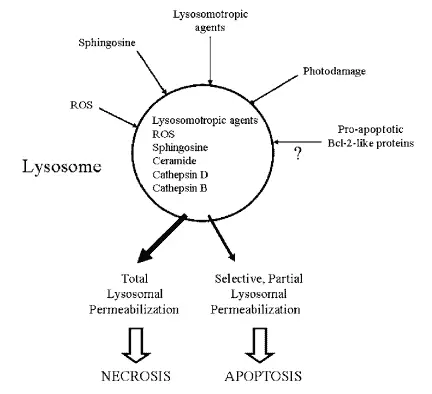
Lysosome enzymes are proteases (catalyzers of protein breakdown) associated with programmed cell death. However, the lysosomal pathway of apoptosis (a form of programmed cell death) is usually related to cases with a previous pathology. Tumorigenic transformation of cells also promotes the activation of the lysosome pathway causing many lysosomal enzymes to be overexpressed in cancer.
In fact, the increased expression of cathepsin B and D have been observed in breast, colorectal, gastric, lung, prostate and thyroid cancers. The cell death pathway can also initiate with the permeabilization (the act of making something permeable) of the lysosome, causing the release of cathepsins and hydrolases from the lysosome to the cytosol. This lysosomal membrane permeabilization is induced by distinct stimuli, such as reacting oxygen species lysosomotropic agents, photodamage, sphingosines, among others.
This process is potentially lethal because some lysosomal proteases cause the digestion of essential proteins in the cytosol and the activation of hydrolases, thus, massive lysosomal breakdown results in cellular necrosis whereas partial and selective lysosomal permeabilization is associated with apoptosis (figure 5) (Guicciardi et al. 2004, Boya & Kroemer 2008).
Lysosomes: Fun Fact
The acid hydrolases can only work in an acidic pH but become inactive at a neutral pH. The fact that cytoplasm has a neutral pH provides protection against uncontrolled digestion of the elements in the cytoplasm, especially in the event that the lysosomal membrane breaks. This mechanism is essential to protect the cell from accidental degradation (Cooper 2000).
Takeaways
To sum up, lysosomes are organelles capable of breaking down molecules with enzymes that work in acidic conditions, known as hydrolases. These organelles are formed by the fusion of clathrin-coated vesicles packaged in the trans-Golgi network with late endosomes.
Lysosomes perform intracellular digestion by receptor-mediated endocytosis, phagocytosis and autophagy to degrade different kinds of elements such as bacteria, cell debris, damaged cells and its own cell’s components. Intracellular digestion is so important to the preservation of the cells that an alteration in any of the aforementioned processes can led to disease. These alterations are in fact known as lysosomal storage diseases and cause more than 30 genetic disorders, have a broad spectrum of symptoms and involved different enzyme and protein activator deficiencies, and the incapability to transport small molecules.
This amazing organelle is can be thought of as the garbage truck of the cell in the overall process of “cell digestion” as it is commonly referred to. Without lysosomes our cells would suffer from the accumulation of molecules that over time would become harmful to the cell and cause a breakdown of the entire cellular structure.
References
- Boya, P., & Kroemer, G. (2008). Lysosomal membrane permeabilization in cell death. Oncogene, 27(50), 6434-6451.
- Cooper, G. M. (2000). The Cell: A Molecular Approach. 2nd edition. Sunderland (MA): Sinauer Associates. Lysosomes.
- Du, H., Cameron, T. L., Garger, S. J., Pogue, G. P., Hamm, L. A., White, E., ... & Grabowski, G. A. (2008). Wolman disease/cholesteryl ester storage disease: efficacy of plant-produced human lysosomal acid lipase in mice. Journal of lipid research, 49(8), 1646-1657.
- Ferreira, C. R., & Gahl, W. A. (2017). Lysosomal storage diseases. Translational science of rare diseases, 2(1-2), 1-71.
- Gartner, L. P., & Hiatt, J. L. (2012). Color atlas and text of histology. Lippincott Williams & Wilkins. 512 pp.
- Greiner-Tollersrud, O. K. & Berg T. (2013). Lysosomal Storage Disorders. In: Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience.
- Guicciardi, M. E., Leist, M., & Gores, G. J. (2004). Lysosomes in cell death. Oncogene, 23(16), 2881-2890.
- Johnson, W. G. (2015). Disorders of glycoprotein degradation: sialidosis, fucosidosis, α-mannosidosis, β-mannosidosis, and aspartylglycosaminuria. In Rosenberg's Molecular and Genetic Basis of Neurological and Psychiatric Disease (pp. 369-383). Academic Press.
- Kishnani, P. S., Steiner, R. D., Bali, D., Berger, K., Byrne, B. J., Case, L. E., ... & Mackey, J. (2006). Pompe disease diagnosis and management guideline. Genetics in Medicine, 8(5), 267-288.
- Lawrence, R. E., & Zoncu, R. (2019). The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol, 21(2), 133-142.
- Maxfield, F. R., Willard, J. M., & Lu, S. (Eds.). (2016). Lysosomes: Biology, Diseases, and Therapeutics. John Wiley & Sons.
- Novikoff, A. B., Beaufay, H., & de Duve, C. (1956). Electron microscopy of lysosome-rich fractions from rat liver. The Journal of biophysical and biochemical cytology, 2(4), 179.
- Pryor, P. R. (2012). Analyzing lysosomes in live cells. In Methods in enzymology (Vol. 505, pp. 145-157). Academic Press.
- Saftig, P. (2005). Lysosomes. Springer Science & Business Media. 197 pp.